Los primeros síntomas del síndrome de Tourette se observan casi siempre a partir de la niñez, iniciándose generalmente entre los 7 y 10 años de edad. El síndrome de Tourette afecta a personas de todos los grupos étnicos, aunque los varones se ven afectados con una frecuencia entre tres o cuatro veces mayor que las mujeres. Se calcula que 200,000 norteamericanos padecen de la forma más severa del síndrome de Tourette mientras que una de cada cien personas presenta síntomas más leves y menos complejos, tales como tics motores o vocales crónicos o los tics pasajeros de la niñez. Aunque el síndrome de Tourette puede manifestarse como condición crónica con síntomas que persisten durante toda la vida, la mayoría de las personas que padecen del mal presentan los síntomas más severos durante los primeros años de adolescencia y van mejorando al avanzar hacia la fase más tardía de la adolescencia y posteriormente en la madurez.
lunes, 17 de enero de 2011
Sindrome de Tourette
Sindrome de asperger
El síndrome de Asperger (SA) es un trastorno del espectro del autista. Es más leve que el autismo pero comparte algunos de sus síntomas. Es más común entre los niños que entre las niñas.
El principal síntoma es un interés obsesivo en un sólo tema. Algunos niños con SA se convierten en especialistas en dinosaurios, marcas y modelos de automóviles, hasta en objetos aparentemente extraños como son las aspiradoras. Sus conocimientos, altos niveles de vocabulario y patrón formal del lenguaje, los hace parecer pequeños profesores.
Los niños con SA tienen dificultad para interpretar situaciones sociales e identificar los sentimientos de otras personas. Pueden tener movimientos extraños o tics nerviosos. Todo esto les dificulta hacer amigos. Los problemas con las habilidades motoras también son comunes en los niños con SA. Por ejemplo, pueden aprender a andar en bicicleta o atrapar una pelota más tarde que otros niños. El tratamiento se enfoca en tres síntomas principales: insuficiencia en las habilidades para comunicarse, rutinas obsesivas o repetitivas y torpeza física.
PROGERIA

Hay 100 casos conocidos en el mundo. Se estima que aparece 1 caso de progeria por cada 8 millones de nacimientos. Este síndrome se produce por mutaciones de herencia autonómica dominante en el gen LMNA. Es una enfermedad genética de la infancia extremadamente rara, caracterizada por un envejecimiento prematuro. Crecen mas lentamente y desarrollan una expresión facial muy característica. A pronta edad, (5-6 años) pierden el pelo, desarrollan arrugas y comienzan a padecer aterosclerosis, como si se tratasen de ancianos, lo que les lleva a la muerte en los primeros años de la adolescencia. La edad de muerte natural sucede a los 12-15 años.
El enanismo.

En la mayoría de los casos el enanismo o acondroplastia es una mutación genética que afecta a una de cada 20.000 personas. La causa principal es un problema en el cartílago, que presenta una mayor concentración de calcio, lo que impide a los huesos crecer a su ritmo normal. Un enano es una persona de baja estatura de menos de 1.25 m en la edad adulta. Más de 200 cuadros distintos pueden causar el enanismo.Un único tipo, llamado acondroplasia, causa casi el 70 por ciento de todos los tipos de enanismo. LA enfermedad tiene lugar cuando los brazos y las piernas se ven cortas en comparación con la cabeza y el tronco. En España existen unas 11.000 personas afectadas por problemas relativos al enanismo, de los que se han distinguido hasta 200 tipos diferentes. Algunos de ellos pueden corregirse mediante actuaciones en la hormona del crecimiento mientras que otros no tienen cura. En cualquier caso, los acondroplásicos requieren cuidados específicos durante toda su vida y, especialmente, durante la niñez ante el riesgo de muerte infantil.


El labio leporino consiste en una hendidura o separación en el labio superior.El labio leporino se origina por fusión incompleta de los procesos maxilar y nasolateral del embrión y es uno de los defectos de nacimiento más frecuentes.El labio leporino es un defecto congénito de las estructuras que forman la boca. Es una hendidura o separación en el labio y es el resultado de que los dos lados del labio superior no crecieron a la vez.
El labio leporino se presenta sobre todo en familias con un historial de esta anormalidad en alguno de los padres, en otro niño o un pariente inmediato. Pero también puede presentar en familias sin los antecedentes ya mencionados.
Durante el desarrollo fetal la boca se forma durante los primeros tres meses del embarazo. Durante este tiempo, las partes del paladar superior y el labio superior normalmente se unen. Cuando la unión no se completa es cuando se presenta en el niño el labio leporino, paladar hendido o ambos.
La signología es por demás obvia, y se detecta inmediatamente al nacimiento.
El niño con labio leporino y/o paladar hendido puede albergar la seguridad de que llegará a hablar, a actuar y a tener un aspecto como todos los demás niños.
Aunque el tratamiento lleva unos cuantos años, vale la pena la espera si se considera el beneficio que puede alcanzarse.
El Síndrome De Dravet
http://www.youtube.com/watch?v=SoC1ba6NfdE
bibliografia:http://dialnet.unirioja.es/servlet/articulo?codigo=647270
www.youtube.com
http://es.wikipedia.org/wiki/S%C3%ADndrome_de_Dravet
domingo, 16 de enero de 2011
El Autismo.

Desde que el Dr. Leo Kanner describió por primera vez el autismo, hace ya más de medio siglo, los investigadores han hecho notables avances en el conocimiento de este trastorno. Hoy, los investigadores se han propuesto una nueva frontera: identificar las bases genéticas del autismo.
La evidencia derivada de muchos estudios sugiere que los factores hereditarios son en gran parte responsables de la ocurrencia de los trastornos relacionados con el autismo. Sin embargo, es igualmente claro que la genética no puede explicar por completo el desarrollo del autismo.
Esto significa que múltiples factores genéticos probablemente están implicados, y pueden predisponer a un individuo a desarrollar el autismo.
El Autismo es un término general utilizado de manera intercambiable ya que los profesionales de la salud llaman Trastornos Generalizados del Desarrollo (TGDs).
Las personas con un trastorno generalizado del desarrollo presentan un conjunto particular de síntomas que afectan tres áreas: comunicación, socialización (interacción con los demás) y comportamiento.
Esta amplia categoría comprende en la actualidad cinco diagnósticos aceptados oficialmente:
.-Trastorno Autista (Autismo)
.-Síndrome de Asperger
.-Trastorno Generalizado del Desarrollo No Especificado
.-Síndrome de Rett
.-Trastorno Desintegrativo Infantil
Las distinciones entre estos diagnósticos se basan en diferencias sutiles en el desarrollo del lenguaje, la severidad del trastorno, y la edad de comienzo (la edad cuando los síntomas aparecen por primera vez), así como en otros aspectos que el profesional de la salud puede evaluar.
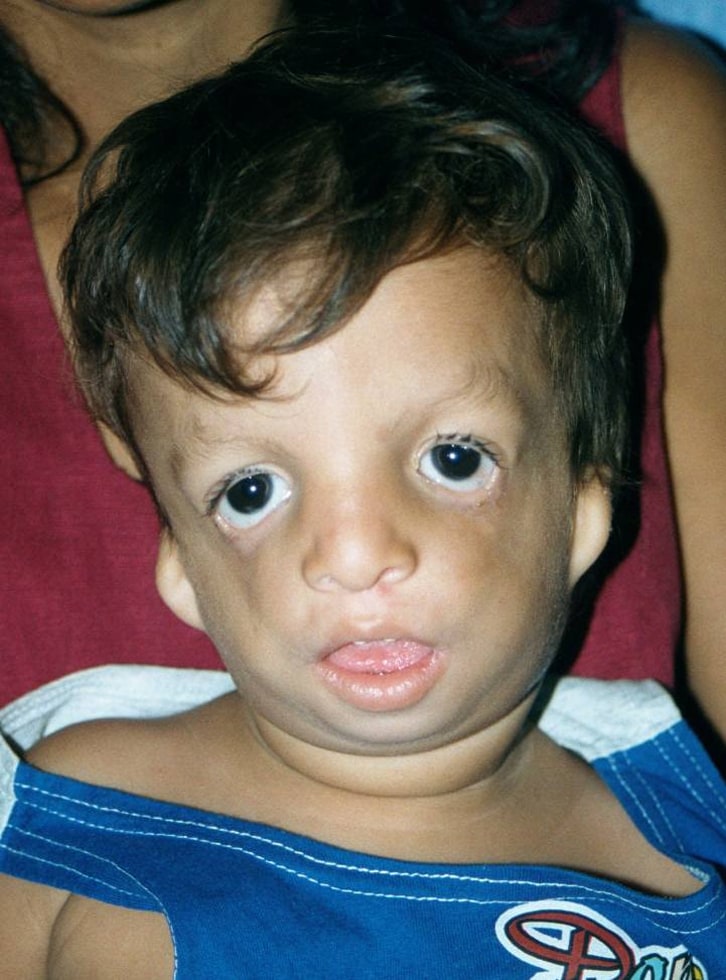
Síndrome de Treacher-Collins
{kind=link}

{kind=link}
Treacher Collins, Franceschetti y otros colaboradores, eran doctores que describieron pacientes con esta apariencia característica en 1900 y 1949 respectivamente. El nombre que el Dr. Franceschetti le dio a esta condición es "disostosis mandibular", lo que simplemente significa que hay un desarrollo anormal de la mandíbula inferior de la cara.